Le site des ophtalmologistes de France
Espace Encyclopédie
Encyclopédie de la vue

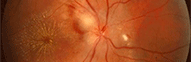




Rétinoblastome
Le rétinoblastome
Retinoblastoma
Nous remercions beaucoup le Dr Laurence Desjardins (Institut Curie Paris France) qui nous propose le chapitre ci-dessous.
Le rétinoblastome est une tumeur hautement maligne touchant essentiellement le nourrisson et le jeune enfant.
C'est une tumeur génétiquement déterminée. Le gène du rétinoblastome siège sur le chromosome 13 q. Pour que la tumeur se développe il faut que les deux allèles du gène soient mutés. Dans le rétinoblastome bilatéral il existe une mutation génétique constitutionnelle présente dans toutes les cellules de l'organisme et la deuxième mutation a lieu au niveau de la cellule rétinienne. C'est pourquoi le gène du rétinoblastome fonctionne comme un gène récessif mais la maladie se transmet selon un mode autausomal dominant. Dans le rétinoblastome unilatéral unifocal non héréditaire dans la plupart des cas les deux mutations ont lieu dans la cellule rétinienne.
Les signes cliniques
Le strabisme peut être un signe d'appel précoce permettant parfois de faire le diagnostic de petites tumeurs se développant dans la région maculaire. C'est pourquoi il ne faut pas confondre le strabisme accommodatif du nourrisson avec un strabisme unilatéral permanent pouvant témoigner d'une atteinte organique de la rétine.
La leucocorie ou reflet blanc dans la pupille est visible au début que sous certains éclairages et dans certaines directions du regard. Puis elle devient permanente. Ces signes doivent impérativement faire pratiquer un examen du fond d'oeil après dilatation.
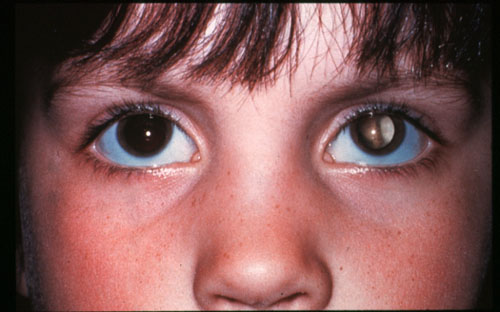
Leucocorie de l'oeil gauche
Les autres signes sont plus tardifs.
On peut citer la buphtalmie, l'hétérochromie irienne, le pseudohypopion et dans les formes les plus évolués l'exophtalmie tumorale.
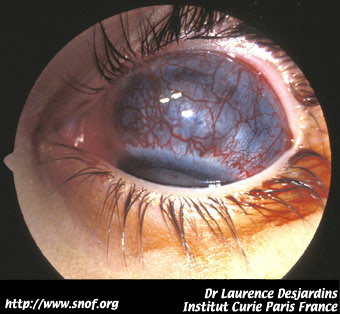
Buphtalmie
Le diagnostic
Il repose sur l'examen du fond d'oeil sous anesthésie générale après dilatation maximum.
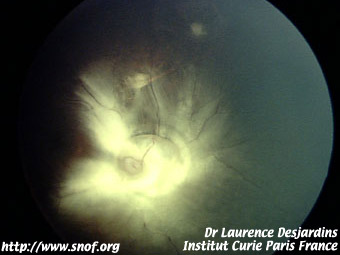
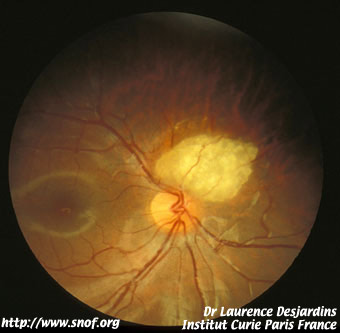
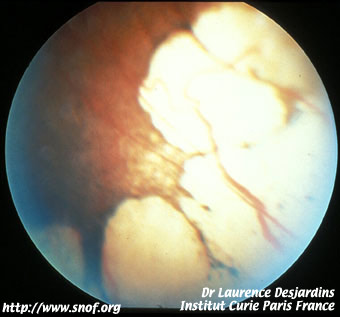
Le rétinoblastome endophytique apparaît comme une tumeur blanche richement vascularisée se développant vers la cavité vitréenne avec de nombreux flocons blanchâtres flottant dans le vitré.
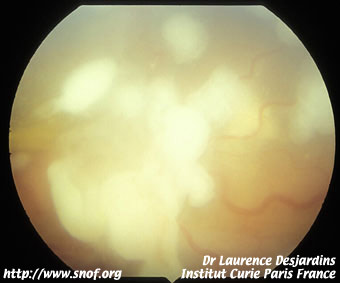
Rétinoblastome endophytique
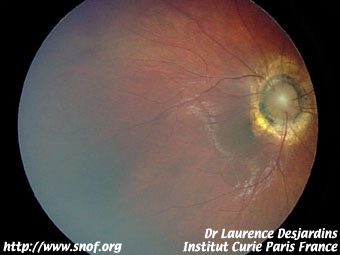
Les formes exophytiques se présentent sous forme d'un décollement de rétine derrière lequel on aperçoit les masses saillantes parfois calcifiées en partie blanches avec dilatation angiomateuse des vaisseaux.
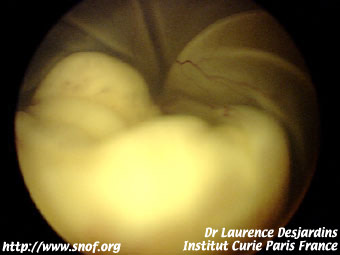
Rétinoblastome exophytique
L'échographie est utile montrant l'extension des lésions avec des masses très échogènes.
Le scanner confirme le diagnostic en montrant des calcifications. Il précise l'envahissement du nerf optique ainsi qu'une éventuelle extension cérébrale.
L'IRM. est souvent préférable chez ces enfants et permet de bien analyser une extension éventuelle au niveau du nerf optique.
Le diagnostic différentiel
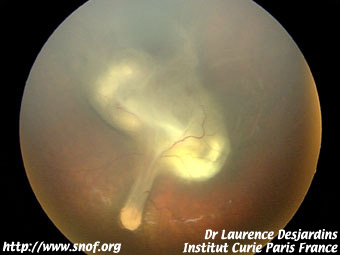
Il se pose surtout avec la maladie de Coats qui est facile à diagnostiquer dans sa forme précoce où il existe des tellangiectasies rétiniennes périphériques caractéristiques; par contre les formes évoluées au stade de décollement total de la rétine, les deux affections peuvent être très difficile à différencier. L'absence de calcification est en principe un signe en faveur de la maladie de Coats.
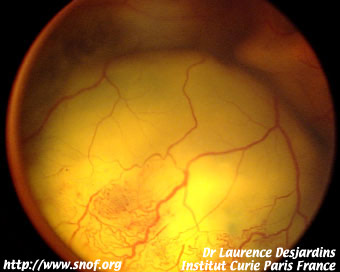
Maladie de Coats
Le rétinoblastome peut être responsable d'une réaction inflammatoire soit au niveau de l'oeil lui-même soit au niveau de l'orbite pouvant orienter à tort vers une uvéite ou une pseudo tumeur inflammatoire.
Il existe une forme de rétinoblastome qui se présente sous forme d'un décollement de rétine ou d'une hyalite sans masse tumorale individualisable ce qui, rend. son diagnostic extrêmement difficile; ainsi le rétinoblastome infiltrant diffus peut ne pas être visible au scanner ou en I.R.M.. En cas de doute il ne faut pas hésiter à adresser l'enfant dans un centre spécialisé. La vitrectomie est particulièrement contre-indiquée en cas de rétinoblastome.
Les autres affections rétiniennes sont en général plus facile à diagnostiquer au fond d'oeil.
Certaines sont des maladies inflammatoires comme le toxocara canis où la maladie des griffes du chat.
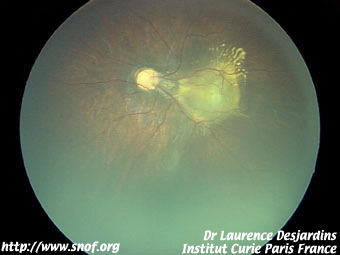 Toxocara canis |
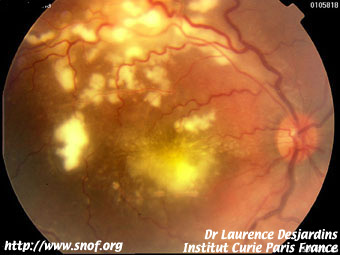 Maladie des griffes du chat |
Certaine sont des affections malformatives comme les fibres à myéline, le morning glory syndrome, la dysplasie rétino vitréenne, les colobomes.
 Fibres à myéline |
 Morning Glory Syndrome |
 Dysplasie rétino-vitréenne |
 Colobome |
Un hamartome combiné est parfois retrouvé:
Hamartome combiné
Des astrocytomes bénins peuvent se voir isolément ou dans le cadre d’une sclérose tubéreuse de Bourneville.
Astrocytome
La prise en charge thérapeutique actuelle
Elle a beaucoup évolué au cours des dix dernières années.
Elle nécessite la collaboration d'une équipe multidisciplinaire habituée au traitement de rétinoblastome.
Les complications de la radiothérapie sont essentiellement la cataracte et la sécheresse oculaire, l'atrophie du massif facial, les insuffisances hypophysaires et surtout les sarcomes secondaires dans le champ d'irradiation. En raison de l'existence de ces complications on essaye actuellement chaque fois que cela est possible d'éviter l’irradiation externe.
La chimiothérapie première par carbo Platine, VP16 et Vincristine permet de diminuer le volume des tumeurs dans le but de les rendre accessibles à des traitements conservateurs autres que l'irradiation externe.
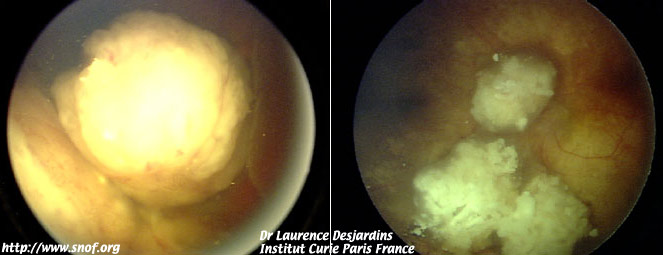
Effets de la chimiothérapie sur un rétinoblastome
Les traitements disponibles et leurs indications :
L'énucléation est indispensable en cas de tumeur massive notamment dans les formes unilatérales.
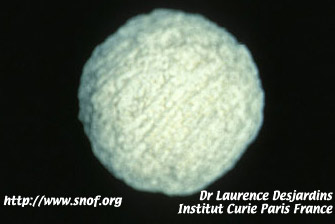
Implant de corail utilisé après énucléation
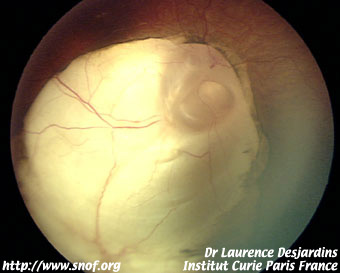
L'énucléation doit être faite par un chirurgien parfaitement entraîné avec section du nerf optique à distance du globe. On utilise actuellement le plus souvent des implants en corail recouverts de treillis de vicryl ou de fascia lata ; l'examen anatomopathologique doit être soigneux. Un traitement complémentaire éventuel chimio ou radiothérapique peut être décidé en fonction des facteurs de risque histologique.
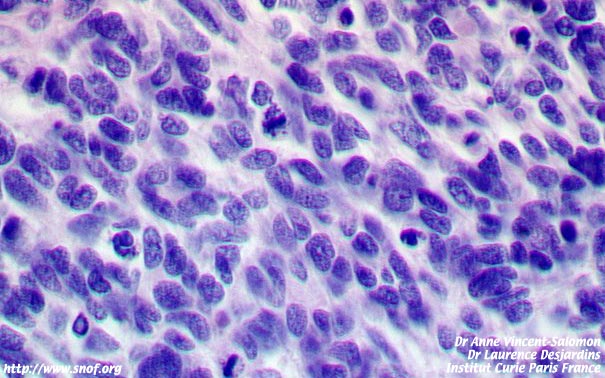
Etude histologique d'un rétinoblastome
Un prélèvement tumoral est en général réalisé en postopératoire immédiat pour des analyses génétiques de la tumeur. L'examen anatomopathologique recherche un envahissement massif de la choroïde, un envahissement rétro laminaire du nerf optique ou un envahissement de la tranche de section voire parfois un envahissement extra scléral.
En accord avec les pédiatres oncologue une chimiothérapie voire parfois une radiothérapie orbitaire sont parfois nécessaires. La radiothérapie orbitaire peut actuellement se faire avec une technique de curiethérapie à l'iode 125 avec des séquelles orbitaires nettement moindres.
La cryoapplication est utilisable pour les petites tumeurs ne dépassant pas 3 mm de diamètre situées au niveau de la rétine périphérique. Elle se fait sous contrôle ophtalmoscopique.Une à trois sessions sont nécessaires pour chaque tumeur.
La curiethérapie à l'iode 125 est indiquée pour les tumeurs périphériques notamment lorsqu'il existe un essaimage localisé dans le vitré. Une dose de 45 grays au sommet est en général suffisante.

Iode 125
Le traitement par thermothérapie au laser diode est efficace pour les petites tumeurs postérieures.Le traitement est réalisé sous anesthésie générale et sous microscope opératoire.
Laser réalisé grâce à un microscope opératoire
La thermochimiothérapie associe une perfusion de Carbo platine dont l'action est potentialisée par une thermothérapie au laser diode. Le laser est réalisé sous anesthésie générale au bloc opératoire dans les deux heures qui suivent la perfusion. C'est actuellement le traitement de choix de la plupart des tumeurs accessibles à un traitement conservateur permettant d'obtenir une guérison dans 90 % des cas.
L'irradiation externe se fait à curie par un faisceau d'électrons avec une dose de 45 grays délivrée à l'ensemble de la rétine. Elle reste indispensable pour les tumeurs massives bilatérales.
Les indications actuelles :
En cas de rétinoblastome unilatéral l'énucléation est indispensable dans 80 % des cas la tumeur étant souvent diagnostiquée un stade tardif. Les formes très évoluées en particulier les buphtalmies où les exophtalmies doivent être d'abord traitées par chimiothérapie avant l'énucléation.
L'examen anatomopathologique est ensuite essentiel.
En cas d’ envahissement massif de la choroïde ou d'envahissement rétrolaminaire du nerf optique un traitement chimiothérapique est nécessaire. En cas d'envahissement de la tranche de section du nerf optique ou d'envahissement extra scléral une radiothérapie orbitaire est en outre indispensable.
Dans 20 % des cas, un traitement conservateur par thermochimiothérapie est possible.
En cas de rétinoblastome bilatéral s’il s’agit d'une forme asymétrique, on réalise en général la l'énucléation de l'oeil le plus atteint et un traitement conservateur sur l'autre œil associant thermochimiothérapie, cryothérapie et disques diode si nécessaire. Les traitements locaux sont en général décidés après une chimiothérapie première pour réduire les volumes tumoraux.
Les résultats visuels sont satisfaisants.
Les yeux qui n'ont pas de cicatrice maculaire ont en général une excellente acuité visuelle.
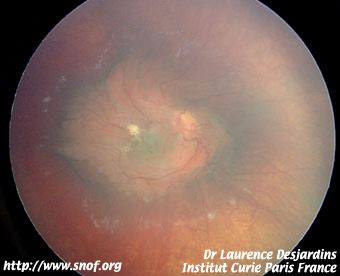
Cicatrices rétiniennes des disques d'iode 125
En cas de rétinoblastome bilatéral évolué les traitements sont également décidés après chimiothérapie première. Il est en général nécessaire d'utiliser une irradiation externe d'un ou des deux yeux. Le traitement conservateur vise à éviter une énucléation bilatérale et à garder une fonction visuelle la meilleure possible.
Les formes extra oculaires et métastatiques se voient surtout dans les pays en voie de développement.
Elles sont gravissimes surtout lorsqu’il existe une atteinte du système nerveux central.
Après la prise en charge thérapeutique initiale une consultation de génétique est souhaitable. Une recherche de mutation est en général réalisée chez l'enfant. La mutation est retrouvée dans la grande majorité des formes bilatérales.
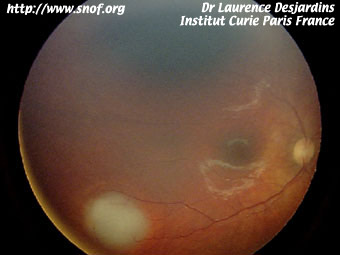
Dépistage d'un rétinoblastome familial
Lorsqu'il existe un antécédent familial de rétinoblastome une consultation de génétique est souhaitable à chaque nouvelle grossesse. Un diagnostic anténatal est parfois possible. Une prise de sang à la naissance permet parfois de savoir si l'enfant est à risque ou non ( lorsque la mutation a été identifiée) Lorsqu'il existe plusieurs cas dans la famille des études génétiques indirectes sont possibles.
Lorsque l'enfant est à risque majeur ( un parent atteint de rétinoblastome unilatéral ou bilatéral, enfant porteur de la mutation ou mutation non identifiée), un suivi mensuel du fond d'oeil dès la naissance est indispensable jusqu'à l'âge de 18 mois et espacé ensuite progressivement. Pendant les trois à quatre premiers mois à l'examen peut se faire sans anesthésie car les tumeurs sont souvent postérieures.Une anesthésie générale et rapidement indispensable à fin de pouvoir dépister les tumeurs périphériques qui peuvent se développer en arrière de l’ora dès trois ou quatre mois de vie. Le diagnostic précoce de la maladie est essentiel et conditionne les résultats visuels chez ces enfants.
En l'absence d'antécédent familial le diagnostic précoce est également essentiel. Il repose sur la bonne connaissance des signes cliniques leucocorie et strabisme qui doivent faire pratiquer un examen du fond d'oeil sans tarder.
Bibliographie
Chapitre rétinoblastome Rapport de la société française d’ophtalmologie 2002 L.Zografos

![]()
Suivez nous sur...
![]()
SNOF
10 rue Schweighaeuser
CS 40028
67080 STRASBOURG Cedex
Tél. 03 88 35 01 09
Fax. 03 88 25 51 90